Quotidiano on line
di informazione sanitaria
Lunedì 25 NOVEMBRE 2024
Friuli Venezia Giuliadi informazione sanitaria
Lunedì 25 NOVEMBRE 2024
Covid. Quale efficacia per gli anticorpi monoclonali di fronte alle varianti? Un nuovo metodo per scoprirlo messo a punto a Trieste
La ricerca è stata condotta da un team dell'Università di Trieste che ha pubblicato il suo lavoro su Scientific Reports (Springer Nature). Analizzati due anticorpi in uso contro il Covid: bamlanivimab e etesevimab. Per il primo le mutazioni più pericolose per la sua efficacia sono quelle siglate E484A/G/K/Q/R/V, Q493K/L/R, S494A/P/R, L452R e F490S, mentre per etesevimab sono K417E/N/T, D420A/G/N, N460I/K/S/T, T415P, e Y489C/S. LO STUDIO.

Gli anticorpi monoclonali sono derivati, tramite particolari procedure di laboratorio, da molecole che il nostro organismo produce naturalmente in risposta ad un’infezione o dopo la somministrazione di un vaccino. I due anticorpi somministrati insieme sono autorizzati dallo scorso febbraio per il trattamento di COVID-19 da lieve a moderato, sia negli Stati Uniti che in Europa.
In particolare, la metodologia applicata in questo studio ha permesso di prevedere e spiegare a livello molecolare gli effetti negativi sull’attività di questi agenti terapeutici di sostituzioni nelle posizioni 452 e 484 di SARS-Cov-2, mutazioni presenti nella ben nota variante delta (indiana).
I risultati principali di questa studio mostrano che, rispetto alla proteina Spike di SARS-CoV-2 nella sua forma wild-type, le mutazioni E484A/G/K/Q/R/V, Q493K/L/R, S494A/P/R, L452R e F490S sono predette come fortemente resistenti nei confronti dell’anticorpo LY-CoV555 (bamlanivimab) mentre le mutazioni K417E/N/T, D420A/G/N, N460I/K/S/T, T415P, e Y489C/S andrebbero a diminuire l’efficacia dell’anticorpo LY-CoV016 (etesevimab)”.
In altri termini per l’anticorpo LY-CoV555 (bamlanivimab) le mutazioni più pericolose per la sua efficacia sono E484A/G/K/Q/R/V, Q493K/L/R, S494A/P/R, L452R e F490S, mentre per l’anticorpo LY-CoV016 (etesevimab) sono K417E/N/T, D420A/G/N, N460I/K/S/T, T415P, e Y489C/S.
In generale, i risultati computazionali presentati, fornendo un razionale molecolare agli effetti delle varianti circolanti di SARS-CoV-2, costituiscono uno strumento rapido e affidabile per identificare i punti di forza e di debolezza delle terapie anticorpali nei confronti di un virus in continua evoluzione fornendo quindi sostanziali informazioni strutturali per lo sviluppo di anticorpi più efficienti.
Figura. Rendering al computer della proteina Spike di SARS-CoV-2 a lunghezza intera, incorporato in un modello di membrana ed in complesso con l'anticorpo monoclonale bamlanivimab (in azzurro).
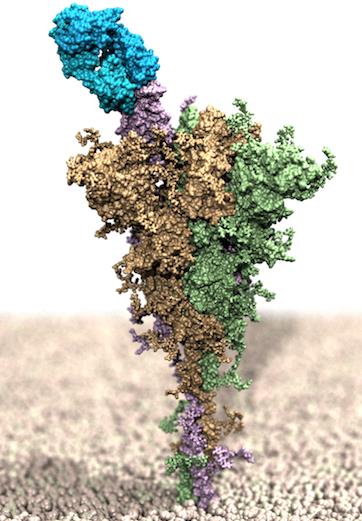
“Rispetto ai precedenti studi eseguiti dal nostro gruppo di ricerca sulle varianti di SARS-CoV-2 – spiega Erik Laurini, co-fondatore del gruppo MolBNL@UniTS – in questo lavoro abbiamo spostato il punto di vista. Prima guardavamo l’effetto di mutanti sull’interazione con la proteina umana che il virus sfrutta per entrare nelle nostre cellule. In quel caso andavamo a prevedere un eventuale aumento della pericolosità e dell’infettività del virus. In questo articolo, invece, siamo andati a valutare il ruolo della diversità genetica virale sull’efficacia di uno degli attuali trattamenti terapeutici. Prevedere in tempi rapidi se una nuova mutazione può compromettere l’efficacia di un agente antivirale sarà fondamentale per combattere definitivamente il COVID-19”.
Per condurre la ricerca sono state adottate delle tecniche di simulazione al calcolatore nell’ambito dell’High Performance Computing (HPC) che hanno permesso l’impiego di risorse e investimenti ridotti, a fronte di un’altissima rapidità di processazione dei dati con tempi non paragonabili a quelli dei laboratori sperimentali.
“Il prossimo obiettivo sarà quello di sfruttare le informazioni raccolte per costruire un modello computazionale in grado di progettare anticorpi monoclonali resistenti alle varianti più pericolose di SARS-Cov-2 - dichiara Domenico Marson, assegnista del gruppo MolBNL@UniTS - Questi farmaci innovativi, infatti, possono essere modificati nella loro struttura in maniera tale da sopperire all’interferenza provocata da una mutazione virale mantenendo allo stesso tempo le altre interazioni che ne garantivano l’efficacia”.
Lo studio, sottolineano i ricercatori, può avere importanti applicazioni nella previsione dell’efficacia di vaccini e terapie come altri tipi di anticorpi monoclonali e farmaci antivirali. L’utilizzo del sistema studiato dal team triestino consentirà di valutare questi e altri rimedi in maniera più veloce ed efficace.
“È importante sottolineare che la procedura computazionale descritta in questo articolo ha un carattere veramente generale. - spiega Sabrina Pricl, coordinatrice del gruppo MolBNL@UnitTS– Infatti, può essere applicata per prevedere in modo rapido ed affidabile l'effetto delle mutazioni anche su altri sistemi di interazione proteina/proteina, così come proteina/ligando e proteina/acido nucleico, che giocano ruoli chiave nella patogenesi di importanti malattie umane tra cui, ad esempio, infezioni batteriche, sindromi ereditarie e, soprattutto il cancro, come già dimostrato in precedenti studi dal nostro gruppo di ricerca. L'HPC, inoltre, è una risorsa strategica per il futuro dell'Europa nel campo delle nanobiotecnologie. Costituisce, infatti, un pilastro dei programmi di finanziamento Europei sia nei precedenti Horizon 2020 che in quelli attuali Horizon Europe”.
21 ottobre 2021
© Riproduzione riservata
Altri articoli in QS Friuli Venezia Giulia






gli speciali

Quotidianosanità.it
Quotidiano online
d'informazione sanitaria.
QS Edizioni srl
P.I. 12298601001
Sede legale e operativa:
Via della Stelletta, 23
00186 - Roma
Quotidiano online
d'informazione sanitaria.
QS Edizioni srl
P.I. 12298601001
Sede legale e operativa:
Via della Stelletta, 23
00186 - Roma
Direttore responsabile
Luciano Fassari
Direttore editoriale
Francesco Maria Avitto
Luciano Fassari
Direttore editoriale
Francesco Maria Avitto
Tel. (+39) 06.89.27.28.41
info@qsedizioni.it
redazione@qsedizioni.it
Coordinamento Pubblicità
commerciale@qsedizioni.it
info@qsedizioni.it
redazione@qsedizioni.it
Coordinamento Pubblicità
commerciale@qsedizioni.it
Copyright 2013 © QS Edizioni srl. Tutti i diritti sono riservati
- P.I. 12298601001
- iscrizione al ROC n. 23387
- iscrizione Tribunale di Roma n. 115/3013 del 22/05/2013
Riproduzione riservata.
Policy privacy
- P.I. 12298601001
- iscrizione al ROC n. 23387
- iscrizione Tribunale di Roma n. 115/3013 del 22/05/2013
Riproduzione riservata.
Policy privacy